graph LR
%% Subgraph A: Standard POA
subgraph A[" "]
node1((A)) -->|Match / Mismatch| node2((T))
node1 -->|Deletion Path Collapsed| node2
%% Label pushed to the bottom of subgraph A
labelA["<b>(a) Standard POA (Ambiguous)</b>"]
end
%% Subgraph B: Skip-Enhanced POA
subgraph B[" "]
node3((A)) -->|Match / Mismatch| node4((T))
node3 -.->|0-cost Skip ε| skip[Skip State]
skip --> node4
%% Label pushed to the bottom of subgraph B
labelB["<b>(b) Skip-Enhanced POA (Resolved)</b>"]
end
%% Styling to make labels look like clean captions and push them down
style labelA fill:none,stroke:none,font-weight:bold
style labelB fill:none,stroke:none,font-weight:bold
style skip stroke-dasharray: 5 5, fill:#e1f5fe, stroke:#0288d1
Introduction
NotePublication Announcement
An expanded version of this work has been peer-reviewed and officially published in IEEE Access. You can access the final, peer-reviewed version of record here: https://doi.org/10.1109/ACCESS.2025.3597547.
In bioinformatics, as well as in other applications of sequence alignment, insertions and deletions can involve fragments of different lengths, raising issues on how and how much to account for their costs. For example, a gap cost purely proportional to the gap length can erroneously discourage long gaps, and on the other hand a low cost for gaps can fragment the alignment into too many short gaps. This has motivated the definitions of various gap penalty functions such as, for example, the affine gap penalty score, introduced for genomic sequences (F.Smith and S.Waterman 1981), that we will use in this post.
Multiple Sequence Alignments (MSA) are typically performed in a progressive manner by iteratively adding a new linear string to the MSA. To this purpose, the latter (the MSA) is often linearized into a lossy representation of the actual similarities and differences of the already aligned strings, and then the iterative, crucial, step of progressive MSA is brought down to a pairwise alignment of the new string against the MSA. This alignment involves two strings of different type, one linear and a more complex one, breaking up the traditional symmetry of pairwise alignments, and therefore suggesting new challenges that include re-opening the issue of how to account for gaps. Indeed, whereas in pairwise alignments an insertion is just the dual of a deletion and their cost is the same, here the insertion can mean two events that are substantially different: wherever the MSA has already located—and already accounted—a gap, then this insertion cost should not be accounted again and again to each new string; on the other hand, when the gap is an insertion that had not occurred yet in the MSA, or a deletion in the new string, then this is a new event, and it has to be penalized as such (L.Loytynoja and N.Goldman 2005; D.-F.Feng and F.Doolittle 1987; G.Higgins. and M.Sharp 1988).
An Elastic Degenerate String (ED-String), denoted \(\widetilde{T}\), is a possible way to compactly represent the outcome of a Multiple Sequence Alignment (MSA) by collapsing common fragments into regular strings, and highlighting gaps and variants as list of strings that can have different size and that can include the empty string (R.Grossi et al. 2017; G.Bernardini et al. 2017, 2020).
For instance, consider the following MSA of three closely-related sequences:
\[ \begin{array}{lllll} \text{AC} & \color{red}\text{TAG} & \text{A} & \color{red}\text{TA} & \text{GG} \\ \text{AC} & \color{red}\text{CA -} & \text{A} & \color{red}\text{AA} & \text{GG} \\ \text{AC} & \color{red}\text{- - -} & \text{A} & \color{red}\text{AT} & \text{GG} \\ \end{array} \]
And its ED-string representation is as follows, where \(\epsilon\) denotes the empty string, and those in curly braces are called degenerate positions:
\[ \widetilde{T} = \textcolor{red}{AC} \Bigg\{ \begin{array}{c} TAG \\ CA \\ \textcolor{red}{\epsilon} \end{array} \Bigg\} \textcolor{red}{A} \Bigg\{ \begin{array}{c} \textcolor{red}{T}\textcolor{blue}{A} \\ AA \\ AT \\ \end{array} \Bigg\} \textcolor{red}{GGGG}\textcolor{green}{C}\textcolor{red}{A} \tag{1}\]
The variants represented by an ED-String introduce a partial order in its letters (as opposed to the total order of letters in a linear string, letters are ordered from left to right, but letters that belong to distinct strings of the same degenerate positions are not comparable), and therefore its edit distance can be computed using Partial Order Alignment (POA) (C.Lee et al. 2002) (as shown in (M.Mwaniki et al. 2022) for the simpler case of D-strings).
However, POA alone does not yield the optimal edit transcript unless the empty string is not properly managed. We solve this problem by adding a new edit event: the skip operation. In this post we suggest a dynamic programming algorithm to optimally align a linear string \(P\) and an ED-String \(\widetilde{T}\) with skips operations.
For example, the optimal alignment of \(P=ACATGGGGAAA\) to the ED-string \(\widetilde{T}\) above involves the letters of \(\widetilde{T}\) highlighted with colors with:
- Red letters being matches.
- A skip operation in that the \(\epsilon\) represents an existing gap locus in the MSA, and hence a free insertion.
- The blue \(A\) is instead a new insertion and therefore it has a cost.
- The green letter \(C\) is a mismatch.
- The last letter of \(P\) has been deleted (at a cost).
Our aim is to compute the correct edit distance (the lowest number of operations needed to convert \(P\) into \(T\) with free cost of skips) and an unambiguous edit transcript (the sequences of actual edit events that convert \(P\) into \(T\) (D.Gusfield 1997)).
For example, the distance between \(P\) and \(\widetilde{T}\) above is 3 (1 insertion, 1 mismatch, 1 deletion), and the edit transcript representing the alignment is MMSMMIMMMMXMD, where M denotes a match, S is a skip, X is a mismatch, and D denotes a deletion.
The notion of ED-strings and, over them, the exact (that is, no edit operations are allowed) matching problem elastic-degenerate string matching (EDSM) problem has attracted some attention in the combinatorial pattern matching community. Since its introduction in (S.Iliopoulos et al. 2017), a series of results have been published both for the exact (R.Grossi et al. 2017; G.Bernardini et al. 2019, 2022; Aoyama et al. 2018) as well as for the approximate version of the problem (G.Bernardini et al. 2020, 2017).
The algorithm we describe here is for a global alignment (the natural choice for the progressive MSA application mentioned above), but its extension to local and semiglobal alignment is straightforward. A semiglobal alignment could be used, for example, in computational pan-genomics, with the ED-string representing a pan-genome (Consortium 2018; M.Alzamel et al. 2018, 2020; V.Carletti et al. 2019) and the pattern being a read to be mapped therein: this is currently one of the most relevant problems in bioinformatics (H.Li et al. 2020; M.Rautiainen and T.Marschall 2020; A.Darby et al. 2020; M.Eizenga et al. 2021; A.Cisłak et al. 2018; A.Cisłak and S.Grabowski 2020).
In Section 3 we explain how we propose to manage skips using the basic case of an elastic but not degenerate strings (which we define in Section 2 with other preliminary definitions), and in Section 4 we extend the idea to ED-strings.
Preliminary Definitions
A string \(X\) is a sequence of elements on an alphabet \(\Sigma\), where the alphabet \(\Sigma\) is a non-empty finite set of letters of size \(|\Sigma|\). We denote with \(\varepsilon\) the empty character (a character whose presence does not change the string it is in), and with \(\epsilon\) the empty string: a string made up of empty characters only.
A string \(\Upsilon\) containing one or more empty characters is an elastic string. The set of all finite strings over an alphabet \(\Sigma\), including the empty string \(\epsilon\) of length \(0\), is denoted by \(\Sigma^*\), while \(\Sigma^+\) denotes the set \(\Sigma^* \setminus \{\epsilon\}\). For any string \(X\), we denote by \(X[i..j]\) the substring of \(X\) that starts at position \(i\) and ends at position \(j\). In particular, \(X[0..j]\) is the prefix of \(X\) that ends at position \(j\), and \(X[i..|X|]\) is the suffix of \(X\) that starts at position \(i\), where \(|X|\) denotes the length of \(X\).
Definition 1 (Elastic Degenerate String (ED-string)) An elastic degenerate string (ED-string) \(\widetilde{S}=\widetilde{S}\{1\}\widetilde{S}\{2\}\dots\widetilde{S}\{n\}\) of length \(n\) over an alphabet \(\Sigma\) is a finite sequence of \(n\) degenerate letters \(\widetilde{S}\{i\}\)’s. Each degenerate letter \(\widetilde{S}\{i\}\) is a finite non-empty set of \(s_i\) strings (also called variants) that can possibly properly include the empty string. We denote with \(\ell_i\) the length of the longest string in \(\widetilde{S}\{i\}\).
When \(s_i=1\), then \(\ell_i=1\) as well, and in that case \(\widetilde{S}\{i\}\) is just a simple letter of \(\Sigma\) and we say that \(\widetilde{S}\{i\}\) is solid, and else we say that it is a degenerate position. The following parameters measure the degeneracy of an ED-string: the total size \(N\) and total width \(W\) of an ED-string \(\widetilde{S}\) are respectively defined as \(N=\sum_{i=1}^{n}s_i \cdot \ell_i\) and \(W=\sum_{i=1}^{n}\ell_i\).
For example, \(\widetilde{T}\) in Equation 1 has length \(n=11\), \(W=14\), and \(N=24\). Moreover, we have that all positions are solid except for \(3\) and \(5\), and hence all \(\ell_i\)’s are equal to \(1\) except \(\ell_3=3\) and \(\ell_5=2\), and therein we have \(s_3=s_5=3\).
Edit Distance with Skips
According to the traditional dynamic programming framework, we incrementally compute the optimal alignments of prefix \(P[0..j]\) of \(P\) against prefix (\(T[0..i]\)) of \(T\) starting with \(i\) and/or \(j\) equal to \(0\) up to the final lengths.
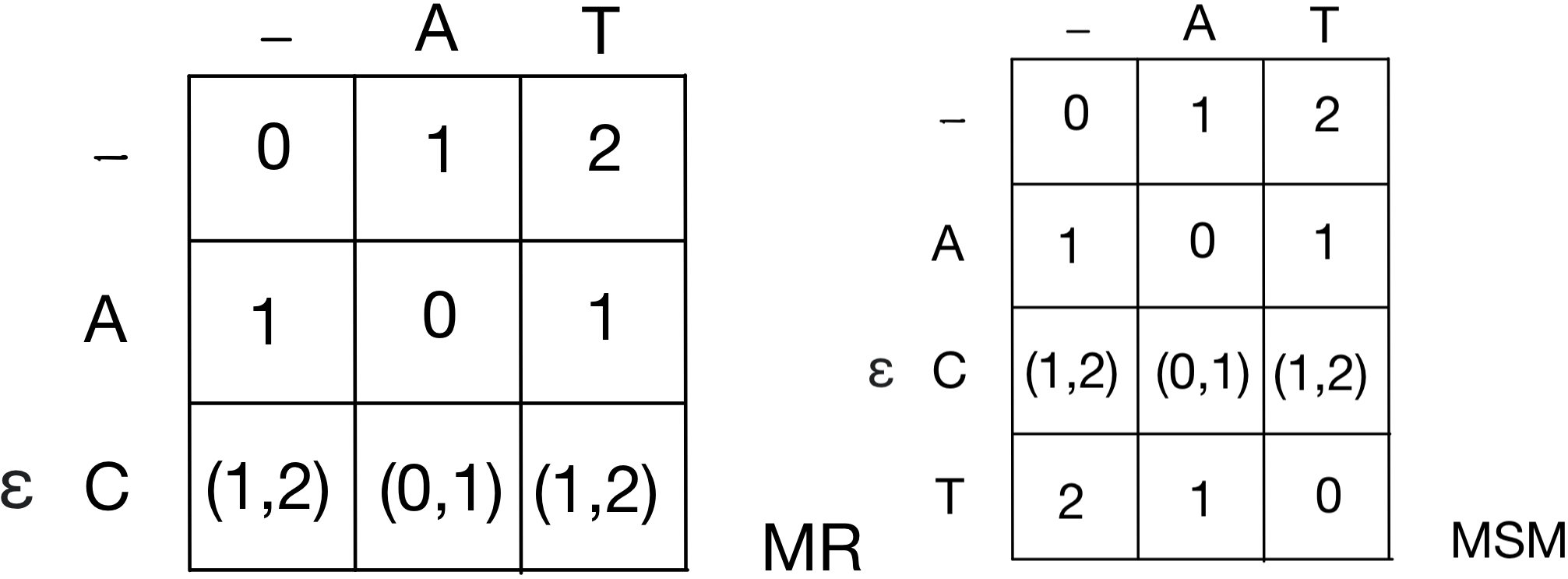
The affine gap penalty function in sequence alignments accounts to a gap of size \(g\) the cost \(w(g)=o+g\cdot e\), where \(o\) is the opening cost, and \(e\) is the extension cost. The problem of computing the optimal alignment with such gap penalty function does not directly fulfill the requirement of having optimal substructure (needed to apply dynamic programming), and to overcome this limitation, the alignment is conceptually split in blocks grouping runs of edit events. To this purpose, the computation of an optimal alignment with affine gap penalty function requires to replicate the matrices: (\(I\)) for insertions, (\(D\)) for deletions, (\(M\)) for (mis)matches. Since the skip operation we want to implement raises a similar issue, we here add an additional matrix (\(S\)) for skips. The skip operation is contemplated when \(\widetilde{T}[i]\) includes the empty string, and it carries a cost of zero because it does not actually change \(P\) and can be thought of as a free insertion.
Let us assume for now that we want to optimally align a string \(P\) with \(|P|=m\) with an elastic string \(\Upsilon \in (\Sigma \cup \varepsilon)^n\) using the scores: \(a\) (match), \(s\) (skip), \(x\) (mismatch), \(o\) (gap opening), \(e\) (gap extend).
We extend the existing model by adding a skip (S) operation that inserts \(\Upsilon[i]\) at no cost when this is an empty character. We therefore end up having edit operations M (for match), X (for mismatch), I (for insertion of \(\Upsilon[i]\neq \varepsilon\)), D (for deletion of \(P[j]\)), and S (for skipping \(\Upsilon[i]=\varepsilon\)), and with the dynamic programming matrices \(M,S,I,D\) of size \((n+1)\times(m+1)\) with the following meanings:
| Matrix | Description |
|---|---|
| \(M[i,j]\) | score of best alignment of \(P[0..j]\) and \(\Upsilon[0..i]\) ending with (mis)match |
| \(S[i,j]\) | score of best alignment of \(P[0..j]\) and \(\Upsilon[0..i]\) ending with a skip of \(\Upsilon[i]\) |
| \(I[i,j]\) | score of best alignment of \(P[0..j]\) and \(\Upsilon[0..i]\) ending with an insertion of \(\Upsilon[i]\) |
| \(D[i,j]\) | score of best alignment of \(P[0..j]\) and \(\Upsilon[0..i]\) ending with a deletion of \(P[j]\) |
The initialization of all matrices is as follows:
- \(M\): Like in traditional dynamic programming with affine gap penalty, we have \(M[0,0]=0\) and \(M[i,0]=M[0,j]= \infty\) because no alignment of \(\Upsilon[0] = \epsilon\) and \(P=\epsilon\) can end with a match or mismatch.
- \(D\): Again like in traditional dynamic programming with affine gap penalty, we have \(D[0, j] = o+e\cdot j\) because we align an empty prefix of \(\Upsilon\) with \(P[0..j]\), while \(D[i,0]=\infty\) because no alignment of \(\Upsilon[0..i]\) and the empty prefix of \(P\) can end with a deletion in \(P\).
- \(I\): Dually, \(I[0,j]=\infty\) because no alignment of an empty prefix of \(\Upsilon\) and \(P[0..j]\) can end with an insertion of a letter of \(\Upsilon\). Let \(i'\) be the smallest \(i\) from \(1\) to \(n\) such that \(\Upsilon[i] \neq \varepsilon\). Then we have:
\[ I[i,0] = \min \begin{cases} \infty & \text{if } \Upsilon[i]= \varepsilon \\ I[i-1,0] + o+e & \text{if } \Upsilon[i] \neq \varepsilon \text{ \& } i=i' \\ I[i-1,0] + e & \text{if } \Upsilon[i] \neq \varepsilon \text{ \& } i\neq i' \\ S[i-1,0]+e & \text{if } \Upsilon[i] \neq \varepsilon \text{ \& } i\neq i' \end{cases} \tag{2}\]
we need to align \(\Upsilon[0..i]\) and the empty prefix of \(P\) ending with an insertion of \(\Upsilon[i]\), and this is only possible if \(\Upsilon[i] \neq \varepsilon\), and it either extends an existing gap that was already open at row \(i-1\), or it extends one that was opened earlier and has included a skip.
- \(S\): We have that \(S[0,j]=\infty\) because no alignment of an empty prefix of \(\Upsilon\) and \(P[0..j]\) can end with a skip of \(\Upsilon\), while:
\[ S[i,0] = \begin{cases} s+\min\{S[i-1,0], I[i-1,0]\} & \text{if } \Upsilon[i]= \varepsilon \\ \infty & \text{if } \Upsilon[i]\neq \varepsilon \end{cases} \tag{3}\]
because we need to align \(\Upsilon[0..i]\) and the empty prefix of \(P\) ending with a skip, and this latter is not possible if \(\Upsilon[i]\neq \varepsilon\). On the other hand, when \(\Upsilon[i]= \varepsilon\), then either a skip had occurred already at the previous row (in which case we inherit its score), or we have to inherit the score of an insertion gap.
We can then progressively fill in the matrices for \(i,j>0\) as follows:
\[ M[i,j] = \min \begin{cases} \infty & \text{if } \Upsilon[i]= \varepsilon \\ f(i,j) + M[i-1,j-1] & \text{if } \Upsilon[i]\neq \varepsilon \\ f(i,j) + S[i-1,j-1] & \text{if } \Upsilon[i]\neq \varepsilon \\ f(i,j) + D[i-1,j-1] & \text{if } \Upsilon[i]\neq \varepsilon \\ f(i,j) + I[i-1,j-1] & \text{if } \Upsilon[i]\neq \varepsilon \end{cases} \tag{4}\]
where:
\[ f(i, j) = \begin{cases} m, & \text{if } \Upsilon[i] = P[j] \\ x, & \text{otherwise} \end{cases} \tag{5}\]
The deletion matrix is the exact same as in traditional alignment:
\[ D[i,j] = \min \begin{cases} D[i, j-1] + e \\ M[i, j-1] + o + e \\ S[i, j-1] + o + e \\ I[i, j-1] + o + e \end{cases} \tag{6}\]
The insertion matrix is as follows:
\[ I[i,j] = \min \begin{cases} \infty & \text{if } \Upsilon[i]= \varepsilon \\ D[i-1, j] + o+ e & \text{if } \Upsilon[i] \neq \varepsilon \\ M[i-1, j] + o + e & \text{if } \Upsilon[i] \neq \varepsilon \\ S[i-1, j] + e & \text{if } \Upsilon[i] \neq \varepsilon \\ I[i-1, j] + e & \text{if } \Upsilon[i] \neq \varepsilon \end{cases} \tag{7}\]
And finally, the skip matrix recurrences:
\[ S[i,j] = \min \begin{cases} s + S[i-1,j] & \text{if } \Upsilon[i]= \varepsilon \\ s + M[i-1,j] & \text{if } \Upsilon[i]= \varepsilon \\ s + I[i-1,j] & \text{if } \Upsilon[i]= \varepsilon \\ s + D[i-1,j] & \text{if } \Upsilon[i]= \varepsilon \\ \infty & \text{if } \Upsilon[i]\neq \varepsilon \end{cases} \tag{8}\]
We then have that the final result is \(\min (M[n, m], I[n, m], D[n, m], S[n, m])\).
Correctness of the Recurrence
Using Figure 1, and given the existing proofs apply for edit distance in general and cases: 1 (alignment ends in an insertion), 2 (alignment ends in a deletion), and 3 (alignment ends in a match or a mismatch) (F.Smith and S.Waterman 1981; O.Gotoh 1982; D.Gusfield 1997).
Lemma 1 When \(\Upsilon[i] = \varepsilon\) we can insert a skip for free.
Proof. In case 4, when \(\Upsilon[i] = \varepsilon\). If the last symbol in the edit transcript is \(S\), inductively the alignment up to \(i-1\) and \(j\) computes the optimal edit distance between \(\Upsilon[i-1]\) and \(P[j]\). Because \(\Upsilon[i] = \varepsilon\), we can insert \(\Upsilon[i]\) after \(P[j]\) without actually changing \(P\), then \(E(i,j) = E[i-1,j]+0\).
Edit Distance with an Elastic Degenerate String
Standard Partial Order Alignment (POA) collapses paths containing deletions, which hides the historical length differences of variants. This causes traceback ambiguity. By introducing a directed “Skip” edge (\(\varepsilon\)), we preserve the exact variant topology without penalizing the alignment.
We can index an MSA column-wise, each column being one base wide, \(0\) being the left-most and \(n\) as the right-most column. After converting this MSA into an ED-String we can apply the same indexing. In this case, a position \(i\) in an ED-String \(\widetilde{T}\), denoted \(\widetilde{T}[i]\) is the set of characters at a locus \(i\) if it were an MSA.
To align a string to an ED-String we pad shorter variants with \(\varepsilon\) until all variants are of equal length.
\[ \widetilde{T} = AC \begin{array}{c} \Bigg\{ \begin{array}{l} TAG \\ C \end{array} \Bigg\} \end{array} A \implies AC \begin{array}{c} \Bigg\{ \begin{array}{c} TAG \\ C\varepsilon\varepsilon\\ \end{array} \Bigg\} \end{array} A \]
The cells of the dynamic programming (DP) table are tuples of size \(\widetilde{T}[i]\) which correspond to the \(E(i,j)\) and functions \(f\) (Equation 5) and \(a\) (which behaves similarly to the skip function in Equation 8) would be extended to Equation 9 and Equation 10.
\[ f(i, j) = \widetilde{T}[i]_{k=0}^{l} : \begin{cases} m, & \text{if } \widetilde{T}[i][k] = P[j] \\ x, & \text{otherwise} \end{cases} \tag{9}\]
\[ a(i, j) = \widetilde{T}[i]_{k=0}^{l} : \begin{cases} s, & \text{if } \widetilde{T}[i][k] = \varepsilon \\ \infty, & \text{otherwise} \end{cases} \tag{10}\]
It then follows that the edit distance to position in a variant is given by \(E(i,j)[k]\). To get the edit transcript we traceback as usual.
Complexity
Given the size of the EDT is \(N\) and the query is \(m\), we have \(O(W \cdot m)\) space and time usage.
Discussion
The introduction of the skip operation (S) resolves a compromise in progressive Multiple Sequence Alignment (MSA) using Partial Order Alignment (POA). While standard POA successfully avoids collapsing alignments into a simple consensus string, its inability to trace distinct variant lengths introduces ambigiuty downstream.
Decoupling Topology from Alignment Cost
The core strength of our approach lies in decoupling the physical topology of the alignment graph from the mathematical cost of path traversal. In standard POA, when variants of different lengths are aligned, the shorter paths are effectively forced to collapse or take penalizing deletions. This creates a highly ambiguous traceback path where true deletions and matches become indistinguishable (as illustrated in Figure 2(a)).
By explicitly mapping empty characters to a dedicated skip matrix (\(S\)) and introducing the zero-cost skip operation (S), we preserve the structural history of the variant. The skipa acts as a topological placeholder: it allows the alignment path to “skip” over pre-existing gap loci without triggering redundant gap-open or gap-extend penalties, while simultaneously generating a precise, unambiguous edit transcript (MMSMMIMMMMXMD).
Practical Trade-offs and Complexity
From a computational complexity standpoint, our algorithm operates in \(O(W \cdot m)\) time and space, where \(W\) is the total width of the ED-String and \(m\) is the query length. This matches the asymptotic bounds of standard pairwise alignment against an ED-String. However, the introduction of the fourth matrix (\(S\)) increases the constant factor of the state space.
In practice, this overhead is a necessary trade-off for obtaining a lossless edit transcript. In large-scale progressive MSA workflows—such as pan-genomic read mapping or deep phylogenetic reconstruction—the lack of an exact traceback transcript compounds error rates with each successive sequence added. Over multiple alignment generations, resolving this transcript ambiguity prevents the “drift” of gap boundaries, yielding significantly cleaner consensus graphs.
SIMD Vectorization Potential
Because our dynamic programming recurrences (Equations Equation 4 to Equation 8) maintain a regular grid-like structure similar to the classic Smith-Waterman-Gotoh formulation, they are highly amenable to parallelization.
Existing high-performance alignment libraries like spoa (R.Vaser et al. 2017) and abPOA (Y.Gao et al. 2020) achieve extreme throughput by using Single Instruction, Multiple Data (SIMD) formulations (specifically, intra-sequence parallelization via SSE or AVX instructions). Our skip recurrences can be mapped to a SIMD layout by treating the skip matrix (\(S\)) updates as parallel vector shifts. Since the skip transition only depends on the state of the previous column (\(i-1\)) when \(\Upsilon[i] = \varepsilon\), the dependency graph remains highly local, allowing vector pipelines to remain fully saturated.
Conclusion and Furture Directions
In this work, we resolved a limitation of Partial Order Alignment (POA): the ambiguity of the edit transcript during progressive Multiple Sequence Alignment. By introducing the skip operation (\(\varepsilon\)) with a zero-cost penalty, we successfully decoupled path traversal from distance calculations. This ensures that existing gaps are not penalized repeatedly while still preserving the exact, unambiguous edit transcript.
Ultimately, viewing an MSA as an Elastic Degenerate String (ED-string) allows for lossless POA with respect to the edit transcript, overcoming a long-standing compromise in progressive sequence alignment.
Future Directions
To build upon these foundational results, we identify two key avenues for future development:
Software Integration and SIMD Acceleration: To make this method viable for high-throughput genomic pipelines, our next step is to implement and optimize the skip-enhanced DP recurrence using SIMD vectorization. This will allow direct integration into state-of-the-art POA libraries like
spoa(R.Vaser et al. 2017) andabPOA(Y.Gao et al. 2020).Application to Large-Scale Progressive MSA: We plan to evaluate the practical impact of lossless traceback transcripts on downstream phylogenetic and variant calling workflows. Quantifying how the elimination of transcript ambiguity improves progressive MSA accuracy across highly polymorphic genomic regions remains a key goal.
References
A.Cisłak, and S.Grabowski. 2020. “SOPanG2: Online Searching over a Pan-Genome Without False Positives.” arXiv:2004.03033 [Cs].
A.Cisłak, S.Grabowski, and J.Holub. 2018. “SOPanG: Online Text Searching over a Pan-Genome.” Bioinformatics 34: 4290–92.
A.Darby, C., R.Gaddipati, M. C.Schatz, and B.Langmead. 2020. “Vargas: Heuristic-Free Alignment for Assessing Linear and Graph Read Aligners.” Bioinformatics 36 (12): 3712–18.
Aoyama, K., Y.Nakashima, T. I, S.Inenaga, H.Bannai, and M.Takeda. 2018. “Faster Online Elastic Degenerate String Matching.” 29th Annual Symposium on Combinatorial Pattern Matching (CPM), LIPIcs, vol. 105.
C.Lee, C.Grasso, and M. F.Sharlow. 2002. “Multiple Sequence Alignment Using Partial Order Graphs.” Bioinformatics 18 (3): 452–64.
Consortium, The Computational Pan-Genomics. 2018. “Computational Pan-Genomics: Status, Promises and Challenges.” Briefings in Bioinformatics 19 (1): 118–35.
D.-F.Feng, and R. F.Doolittle. 1987. “Progressive Sequence Alignment as a Prerequisitet to Correct Phylogenetic Trees.” Journal of Molecular Evolution 25 (4): 351–60.
D.Gusfield. 1997. Algorithms on Strings, Trees, and Sequences: Computer Science and Computational Biology. Cambridge University Press.
F.Smith, T., and M. S.Waterman. 1981. “Identification of Common Molecular Subsequences.” Journal of Molecular Biology 147 (1): 195–97.
G.Bernardini, N.Pisanti, S. P.Pissis, and G.Rosone. 2017. “Pattern Matching on Elastic-Degenerate Text with Errors.” 24th Int. Symposium on String Processing and Information Retrieval (SPIRE), Springer LNCS, vol. 10508: 74–90.
G.Bernardini, N.Pisanti, S. P.Pissis, and G.Rosone. 2020. “Approximate Pattern Matching on Elastic-Degenerate Text.” Theoretical Computer Science 812: 109–22.
G.Bernardini, P.Gawrychowski, N.Pisanti, S. P.Pissis, and G.Rosone. 2019. “Even Faster Elastic-Degenerate String Matching via Fast Matrix Multiplication.” 46th International Colloquium on Automata, Languages, and Programming (ICALP), LIPIcs, vol. 132: 21:1–15.
G.Bernardini, P.Gawrychowski, N.Pisanti, S. P.Pissis, and G.Rosone. 2022. “Elastic-Degenerate String Matching via Fast Matrix Multiplication.” SIAM Journal of Computing 51 (3): 549–76.
G.Higgins., D., and P. M.Sharp. 1988. “CLUSTAL: A Package for Performing Multiple Sequence Alignment on a Microcomputer.” Gene 73 (1): 237–44.
H.Li, X.Feng, and C.Chu. 2020. “The Design and Construction of Reference Pangenome Graphs with Minigraph.” Genome Biology 21 (265).
L.Loytynoja, A., and N.Goldman. 2005. “An Algorithm for Progressive Multiple Alignment of Sequences with Insertions.” Proceedings of the National Academy of Science 102 (30).
M.Alzamel, L. A. K.Ayad, G.Bernardini, et al. 2018. “Degenerate String Comparison and Applications.” 18th International Workshop on Algorithms in Bioinformatics (WABI), LIPIcs, vol. 113: 21:1–14.
M.Alzamel, L. A. K.Ayad, G.Bernardini, et al. 2020. “Comparing Degenerate Strings.” Fundamenta Informaticae 175 (1-4): 41–58.
M.Eizenga, J., A. M.Novak, E.Kobayashi, et al. 2021. “Efficient Dynamic Variation Graphs.” Bioinformatics 36 (21): 5139–44.
M.Mwaniki, N., E.Garrison, and N.Pisanti. 2022. “Fast Exact String to d-Texts Alignments.” CoRR abs/2206.03242.
M.Rautiainen, and T.Marschall. 2020. “GraphAligner: Rapid and Versatile Sequence-to-Graph Alignment.” Genome Biology 21 (253).
O.Gotoh. 1982. “An Improved Algorithm for Matching Biological Sequences.” Journal of Molecular Biology 162 (3): 705–8.
R.Grossi, C. S.Iliopoulos, C.Liu, et al. 2017. “On-Line Pattern Matching on Similar Texts.” 28th Annual Symposium on Combinatorial Pattern Matching (CPM), LIPIcs, vol. 78: 9:1–14.
R.Vaser, I.Sović, N.Nagarajan, and M.Šikić. 2017. “Fast and Accurate de Novo Genome Assembly from Long Uncorrected Reads.” Genome Research 27 (5): 737–46.
S.Iliopoulos, C., R.Kundu, and S. P.Pissis. 2017. “Efficient Pattern Matching in Elastic-Degenerate Texts.” 11th International Conference on Language and Automata Theory and Applications (LATA), Springer LNCS, vol. 10168: 131–42.
V.Carletti, P.Foggia, E.Garrison, L.Greco, P.Ritrovato, and M.Vento. 2019. “Graph-Based Representations for Supporting Genome Data Analysis and Visualization: Opportunities and Challenges.” 12th International Workshop on Graph-Based Representations in Pattern Recognition (GbRPR), Springer LNCS, vol. 11510: 237–46.
Y.Gao, Y.Liu, Y.Ma, B.Liu, Y.Wang, and Y.Xing. 2020. “abPOA: An SIMD-Based C Library for Fast Partial Order Alignment Using Adaptive Band.” bioRxiv.